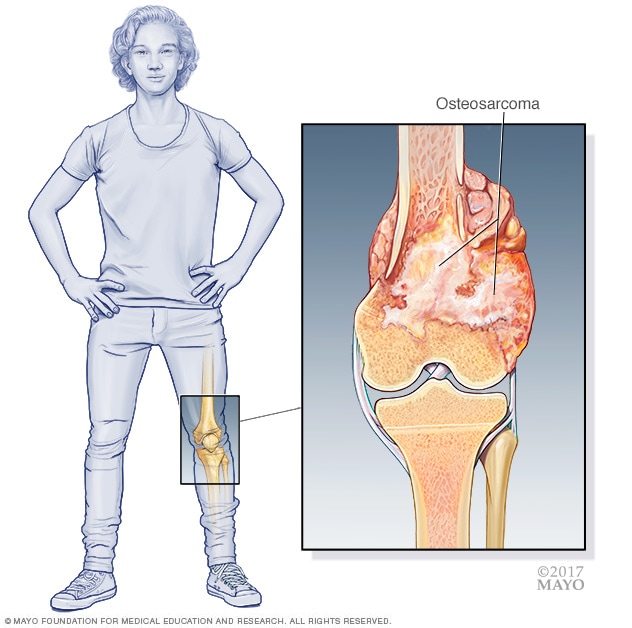
Osteogenic sarcoma, commonly referred to as osteosarcoma, stands as the most prevalent primary malignant bone tumor, predominantly affecting adolescents and young adults. This aggressive form of cancer originates in osteoblasts – the cells responsible for forming new bone tissue – leading to the uncontrolled proliferation of immature bone cells that produce osteoid (unmineralized bone matrix). While it can develop in any bone, it most frequently occurs in the long bones of the limbs, particularly around the knee (distal femur, proximal tibia) and the upper arm (proximal humerus). Understanding osteosarcoma is crucial for early diagnosis and effective management, which often involves a multidisciplinary approach combining surgery, chemotherapy, and sometimes radiation. Its complex biological nature and the significant impact it has on patients’ lives underscore the ongoing need for advanced research and innovative therapeutic strategies.
Understanding Osteogenic Sarcoma: A Comprehensive Overview
Osteosarcoma is characterized by the direct formation of immature bone by the malignant stromal cells. This distinguishes it from other bone tumors, such as Ewing sarcoma, which involves a different cellular origin. The peak incidence of osteosarcoma occurs during periods of rapid bone growth, typically between the ages of 10 and 20, coinciding with adolescent growth spurts. A second, smaller peak is observed in older adults, often in association with pre-existing conditions like Paget’s disease of bone, prior radiation therapy, or inherited cancer syndromes.
Classification and Subtypes
Osteosarcoma is a heterogenous disease, encompassing several histological subtypes, each with unique microscopic features and clinical behaviors. The most common form, accounting for approximately 75-80% of all osteosarcomas, is the conventional (or classical) osteosarcoma. This subtype is further categorized based on its predominant matrix production, such as osteoblastic, chondroblastic, and fibroblastic types.
- Conventional Osteosarcoma: Typically high-grade, meaning it grows rapidly and has a high potential for metastasis. It usually arises within the medullary cavity of the bone.
- Osteoblastic: Characterized by abundant osteoid production.
- Chondroblastic: Features a significant cartilaginous component in addition to osteoid.
- Fibroblastic: Dominated by spindle cell stroma and collagen production, with lesser osteoid.
- Telangiectatic Osteosarcoma: A rare and highly aggressive variant characterized by large blood-filled cystic spaces, often mistaken for benign lesions.
- Small Cell Osteosarcoma: Another rare subtype composed of small, round cells, posing diagnostic challenges due to its resemblance to other small round cell tumors.
- Low-Grade Central Osteosarcoma: A less aggressive form that grows slowly within the medullary cavity.
- Surface Osteosarcomas: These arise on the surface of the bone and include parosteal, periosteal, and high-grade surface osteosarcomas, differing in their aggressiveness and prognosis.
- Parosteal Osteosarcoma: Typically low-grade, arising from the outer surface of the bone, often with a better prognosis.
- Periosteal Osteosarcoma: Intermediate-grade, originating from the periosteum, with a cartilaginous matrix.
The specific subtype plays a critical role in determining treatment strategies and predicting patient outcomes. High-grade osteosarcomas carry a higher risk of local recurrence and distant metastasis, primarily to the lungs.
Etiology and Risk Factors: Unraveling the Causes
The exact cause of osteosarcoma remains largely unknown, making it challenging to establish definitive preventative measures. However, a combination of genetic, environmental, and developmental factors is believed to contribute to its pathogenesis. The rapid bone growth during adolescence is a significant physiological factor, suggesting a vulnerability during intense cellular activity.
Genetic Predisposition
While most cases of osteosarcoma are sporadic, a small percentage are associated with inherited genetic mutations. These germline mutations often increase an individual’s susceptibility to developing the disease.
- Retinoblastoma (RB1) Gene Mutation: Individuals with hereditary retinoblastoma, caused by a mutation in the RB1 tumor suppressor gene, have a significantly elevated risk of developing osteosarcoma, often at a younger age and in multiple bones.
- Li-Fraumeni Syndrome (TP53 Gene Mutation): This rare inherited disorder, characterized by mutations in the TP53 tumor suppressor gene, predisposes individuals to a wide range of cancers, including osteosarcoma.
- Rothmund-Thomson Syndrome: A rare autosomal recessive disorder associated with specific bone abnormalities and an increased risk of osteosarcoma.
- Bloom Syndrome: Another rare genetic disorder that increases the risk of various cancers, including osteosarcoma.
- Werner Syndrome: A disorder causing premature aging and an increased risk of sarcomas.
Environmental and Other Factors
Exposure to certain environmental factors or pre-existing conditions can also increase the risk, though these account for a smaller proportion of cases.
- Previous Radiation Exposure: High-dose radiation therapy, particularly for other cancers, can induce secondary osteosarcoma years or even decades after treatment. The risk is dose-dependent and related to the field of radiation.
- Paget’s Disease of Bone: This chronic bone disorder, more common in older adults, leads to abnormal bone remodeling. In a small percentage of cases, Paget’s disease can transform into osteosarcoma.
- Bone Infarcts and Chronic Osteomyelitis: While less common, these conditions, characterized by areas of dead bone tissue or persistent bone infection, have been implicated as potential risk factors.
- Foreign Bodies: Rarely, metallic implants or other foreign materials left in the bone after injury or surgery have been associated with osteosarcoma development.
It’s important to note that the vast majority of individuals with these risk factors will not develop osteosarcoma, highlighting the complex interplay of genetic and environmental influences.
Clinical Presentation and Diagnostic Pathways
The symptoms of osteosarcoma can be subtle in its early stages, often leading to delays in diagnosis. Awareness of the typical presentation is crucial for healthcare professionals.

Common Symptoms
- Pain: The most common symptom, often localized to the affected bone. Initially, the pain may be intermittent and mild, sometimes attributed to sports injuries or growing pains in adolescents. As the tumor progresses, the pain becomes more persistent, severe, and may worsen at night or with activity.
- Swelling or Lump: A palpable mass or swelling around the affected area may develop as the tumor grows. This swelling can sometimes be accompanied by warmth or tenderness.
- Limited Range of Motion: If the tumor is near a joint, it can cause stiffness and reduce the joint’s mobility.
- Limping: When the tumor affects a leg bone, it can cause a noticeable limp.
- Pathological Fracture: In some cases, the tumor weakens the bone to such an extent that a fracture occurs with minimal trauma. This is often the first symptom that leads to a diagnosis.
Diagnostic Process
A thorough diagnostic workup is essential for confirming osteosarcoma, determining its extent (staging), and guiding treatment.
- Imaging Studies:
- X-rays: Typically the first imaging test performed. Osteosarcoma usually appears as an aggressive lesion with bone destruction, irregular new bone formation (osteoid matrix), and often a soft tissue mass. Characteristic signs include a “sunburst” pattern or Codman’s triangle.
- Magnetic Resonance Imaging (MRI): Provides detailed images of the tumor’s extent within the bone and into surrounding soft tissues, crucial for surgical planning. It can also assess skip lesions (smaller, separate tumor foci within the same bone).
- Computed Tomography (CT) Scan: Used for precise evaluation of bone destruction and matrix mineralization. A CT scan of the chest is mandatory to check for lung metastases, which are common.
- Positron Emission Tomography (PET) Scan: May be used to detect distant metastases throughout the body and assess response to chemotherapy.
- Bone Scintigraphy (Bone Scan): Uses a radioactive tracer to identify areas of increased bone activity, helping to detect other bone lesions or metastases.
- Biopsy: A definitive diagnosis requires a biopsy, where a small tissue sample is extracted from the tumor for pathological examination. This is a critical step, and the biopsy must be performed by an experienced surgeon to avoid compromising future limb-salvage surgery.
- Core Needle Biopsy: A less invasive method using a needle to extract tissue cores.
- Open Biopsy: A surgical procedure to remove a larger piece of tissue.
The biopsy confirms the presence of malignant osteoblasts and allows for histological subtyping and grading of the tumor. Staging involves determining if the cancer has spread from its primary site. Osteosarcoma is generally staged using a system that considers the tumor’s grade (how aggressive it looks under a microscope), its local extent, and the presence of metastases.
Treatment Modalities and Prognostic Considerations
Treating osteosarcoma requires a highly coordinated, multidisciplinary approach involving orthopedic oncologists, medical oncologists, radiation oncologists, pathologists, radiologists, and rehabilitation specialists. The goal is to eradicate the tumor, preserve limb function, and prevent recurrence and metastasis.
Treatment Strategies
- Neoadjuvant Chemotherapy: This involves chemotherapy administered before surgery. Its primary goals are to shrink the tumor, kill micrometastases (tiny undetected cancer cells that may have already spread), and assess the tumor’s response to treatment. Common chemotherapy drugs include doxorubicin, cisplatin, methotrexate (high-dose), and ifosfamide.
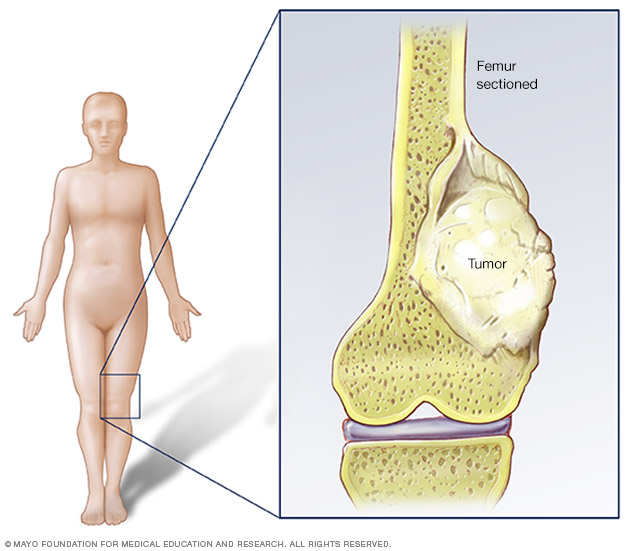
- Surgery: The cornerstone of treatment. For localized disease, the primary tumor is surgically removed.
- Limb-Salvage Surgery: In the majority of cases, advancements in surgical techniques and prosthetic implants allow for removal of the cancerous portion of the bone while preserving the limb. The resected bone is typically replaced with a custom-made metal prosthesis, an allograft (bone from a donor), or a combination.
- Amputation: In some cases, such as when the tumor involves critical nerves or blood vessels, or if the limb cannot be reconstructed functionally, amputation may be necessary.
- Adjuvant Chemotherapy: Administered after surgery to kill any remaining cancer cells and reduce the risk of recurrence and metastasis. The specific regimen depends on the tumor’s response to neoadjuvant therapy and other prognostic factors.
- Radiation Therapy: While osteosarcoma is generally not highly sensitive to radiation, it may be used in specific situations:
- To control unresectable tumors (tumors that cannot be surgically removed).
- For palliative care to relieve pain from metastases.
- In cases where surgical margins are close or positive, to reduce local recurrence risk, although its efficacy in this setting is debated.
Prognosis and Follow-up
The prognosis for osteosarcoma has significantly improved over the past few decades due to the advent of effective chemotherapy. The main prognostic factors include:
- Presence of Metastasis at Diagnosis: Patients with metastatic disease at presentation have a significantly worse prognosis. The lungs are the most common site of metastasis.
- Tumor Response to Neoadjuvant Chemotherapy: A good histological response (e.g., >90% tumor necrosis) to pre-operative chemotherapy is a strong predictor of better outcomes.
- Surgical Margins: Achieving wide, clear surgical margins (no cancer cells at the edge of the removed tissue) is crucial for preventing local recurrence.
- Tumor Size and Location: Larger tumors and those in anatomically challenging locations may have a poorer prognosis.
- Histological Subtype: Low-grade tumors generally have a better prognosis than high-grade conventional osteosarcoma.
Following treatment, patients require diligent long-term follow-up with regular imaging (chest X-rays/CT scans, local MRI) to monitor for recurrence or new metastases. Rehabilitation is also vital to help patients regain strength and function after surgery.
The Future of Osteosarcoma Management
Despite significant progress, osteosarcoma remains a formidable challenge, particularly for patients with metastatic or recurrent disease. Ongoing research aims to improve outcomes through various avenues.
Advances in Research and Treatment
- Targeted Therapies: Scientists are exploring drugs that specifically target molecular pathways involved in osteosarcoma growth and survival. This includes investigating inhibitors of signaling pathways (e.g., mTOR, IGF-1R, angiogenesis inhibitors) that are often dysregulated in osteosarcoma cells.
- Immunotherapy: Harnessing the body’s immune system to fight cancer is a promising area. Clinical trials are investigating checkpoint inhibitors, adoptive cell therapies, and oncolytic viruses for osteosarcoma.
- Precision Medicine: Genetic profiling of individual tumors can help identify specific mutations or biomarkers, allowing for personalized treatment strategies tailored to a patient’s unique cancer.
- Improved Imaging Techniques: Developing more sensitive imaging modalities for earlier detection of recurrence and metastasis.
- Novel Drug Delivery Systems: Research into nanoparticles and other advanced delivery methods to ensure higher drug concentrations reach the tumor while minimizing systemic toxicity.
- Minimally Invasive Surgery and Reconstruction: Continued innovation in surgical techniques and biomaterials for limb reconstruction aims to improve functional outcomes and reduce morbidity.
Collaborative efforts among researchers, clinicians, and patient advocacy groups are driving the quest for more effective and less toxic treatments for osteosarcoma, ultimately offering hope for better long-term survival and quality of life for affected individuals.