The Foundation of Medical Device Classification: A Cornerstone for Tech Adoption
In the rapidly evolving landscape of technological innovation, particularly in sectors that intersect with human health, understanding regulatory frameworks is paramount. A critical first step for any innovator looking to bring a product to market, especially one with a medical application, is to grasp the concept of device classification. Among these classifications, Class I medical devices represent the foundational tier, characterized by their minimal risk and the most streamlined regulatory pathway.
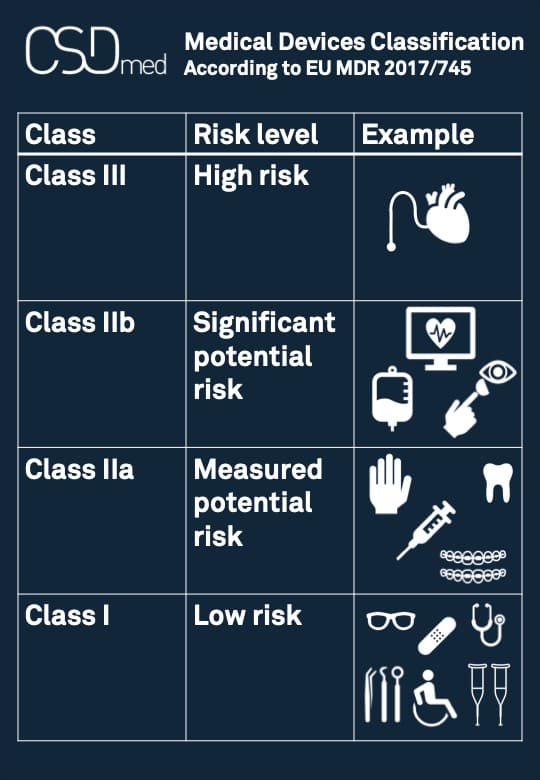
The Food and Drug Administration (FDA) in the United States, along with similar regulatory bodies globally, employs a risk-based classification system for medical devices. This system categorizes devices into three classes (I, II, and III) based on the level of control necessary to assure their safety and effectiveness. Class I devices are specifically designated as posing the lowest potential risk to the patient and/or user. Consequently, they are subject to the least stringent regulatory oversight, primarily focusing on what are known as “general controls.” These general controls are a set of baseline requirements applicable to all medical devices, regardless of class, ensuring basic safety and quality. They include provisions for good manufacturing practices (GMP), proper labeling, adverse event reporting, and registration of manufacturers and devices.
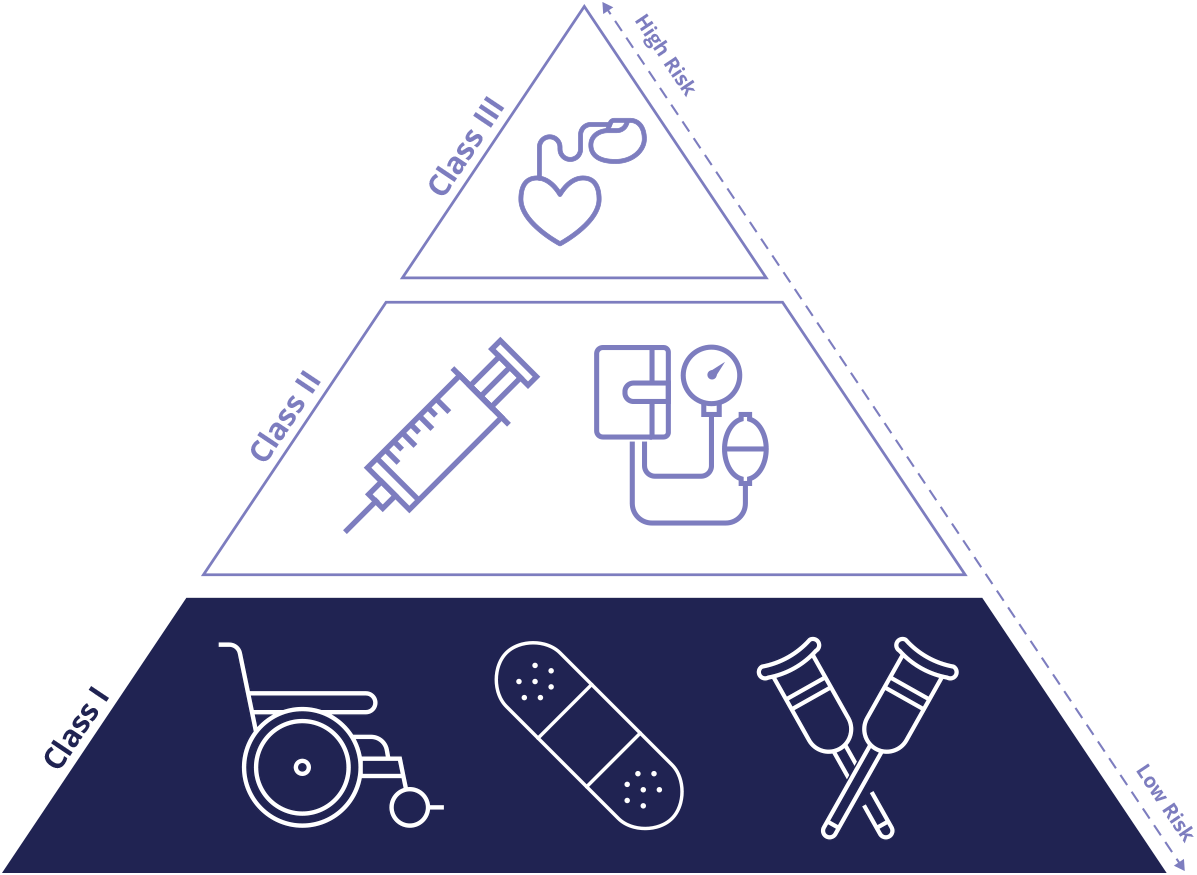
Examples of Class I medical devices illustrate their inherent low risk. These can range from simple items like elastic bandages, tongue depressors, and examination gloves to certain types of dental floss, manual stethoscopes, and some basic surgical instruments. Even some hospital beds, if their intended use is for general support and not for specific therapeutic or diagnostic purposes, can fall into this category. What unites these diverse products is that their potential for harm is low, and their safety and effectiveness can be adequately assured by adhering to the foundational general controls without requiring extensive premarket review or clinical trials. This classification acknowledges that not all technologies present the same level of risk, allowing for an efficient regulatory approach that matches the scrutiny to the potential impact.
Defining Risk and Regulation in Innovation
The core principle behind Class I designation is a careful assessment of risk. Innovators in any field, from advanced robotics to sophisticated software, must consider the potential hazards their products might introduce. For medical devices, this risk assessment dictates the entire development and market entry strategy. A Class I designation signifies that the known and potential risks associated with the device are minimal and can be sufficiently mitigated through fundamental quality assurance and clear instructions for use.
Crucially, “low risk” does not equate to “no risk.” Every medical device carries some inherent degree of risk. However, for Class I devices, these risks are typically well-understood, easily manageable, and unlikely to cause serious injury or illness. For instance, an elastic bandage, while generally safe, could cause skin irritation if applied incorrectly or if the user has an allergy to its material. The general controls address such scenarios by requiring clear labeling, proper manufacturing, and complaint handling systems. This balanced approach allows for faster public access to essential, low-risk medical tools while maintaining a baseline of safety and accountability across the entire medical device industry. For the burgeoning world of tech innovation, understanding this fundamental tier is the first step in responsibly developing and deploying technologies that might eventually touch healthcare.
Navigating Innovation with Minimal Regulatory Hurdles
For innovators, the Class I designation represents a significant advantage, often allowing for a more agile and less resource-intensive path to market. This streamlined process is a deliberate design feature of the regulatory system, intended to avoid stifling the development and adoption of low-risk, yet often highly beneficial, medical technologies.
Accelerating Market Entry for Essential Technologies
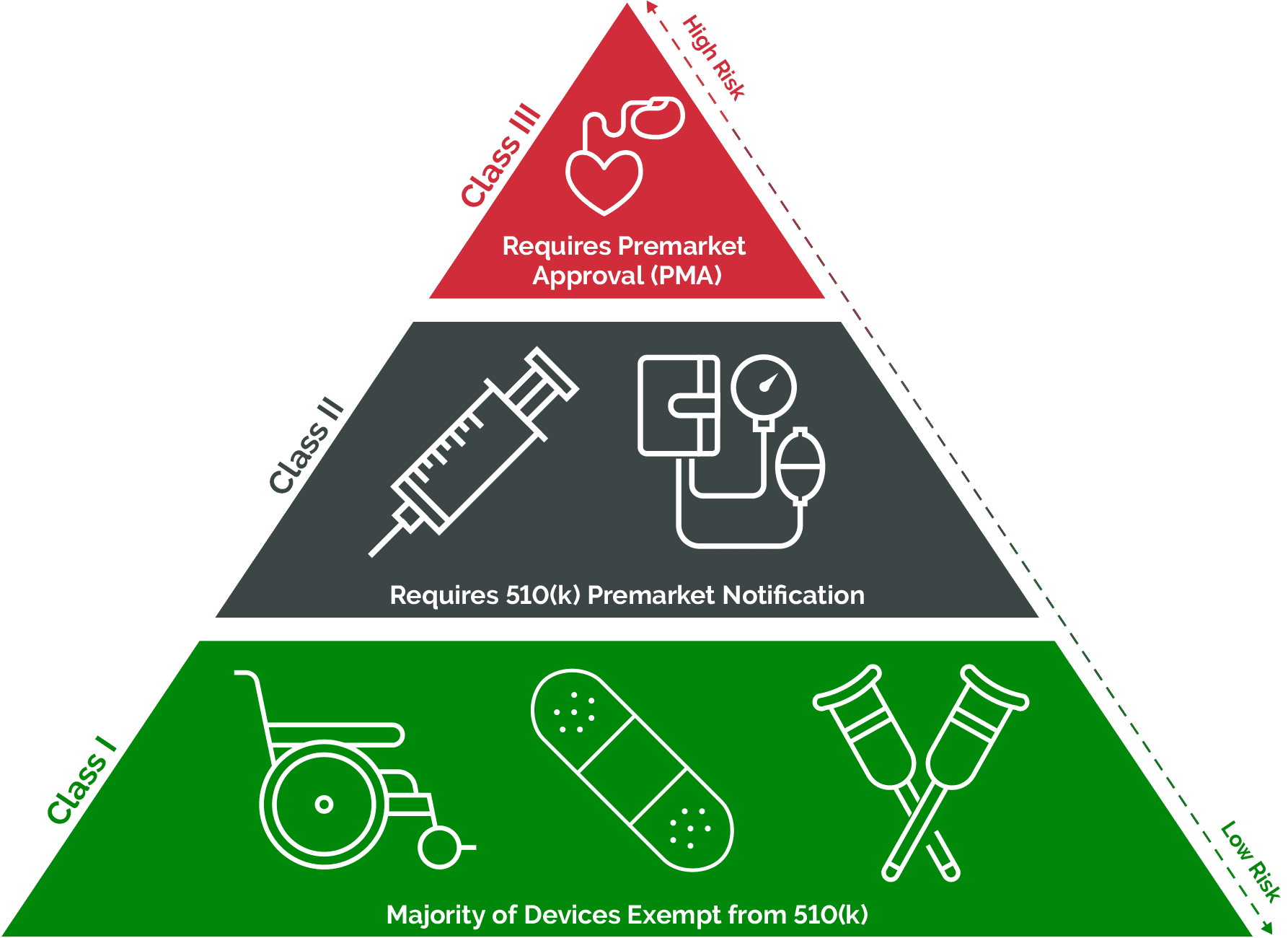
The most direct benefit of a Class I classification is the accelerated market entry. Unlike Class II and particularly Class III devices, which require extensive premarket submissions (like 510(k) clearances or premarket approval (PMA) applications) involving detailed testing, performance data, and often costly and time-consuming clinical trials, most Class I devices are exempt from premarket notification. This means that once a manufacturer ensures their product meets the general controls, they can typically bring it to market without prior FDA review. This exemption significantly reduces development costs and timelines, making it feasible for smaller startups and individual innovators to introduce new, low-risk solutions.
For technologies at the cutting edge, such as certain basic sensors or data collection tools that might eventually integrate into larger health systems, understanding this classification can guide product development. Focusing on functionalities that inherently fall into Class I can enable faster iteration and proof-of-concept, allowing innovators to gather user feedback and refine their products without being bogged down by complex regulatory demands from the outset. This agility is crucial in dynamic tech environments where speed to market can determine success. The emphasis remains on general controls: robust quality management systems, accurate labeling, stringent manufacturing practices, and a system for tracking and reporting adverse events, even for the simplest devices. These foundational requirements ensure that even with minimal premarket review, a safety net is in place.
The Landscape of Low-Risk Digital Health and Emerging Tech
The digital health revolution has introduced a new frontier for medical devices, and many software as a medical device (SaMD) applications or simple data-gathering tools can, in fact, be categorized as Class I. For instance, a mobile application that merely tracks calorie intake or exercise without providing diagnostic or treatment recommendations could be considered a Class I device, or even fall outside medical device regulation entirely depending on its claims. Similarly, basic health information displays or patient engagement tools that do not involve complex algorithms for diagnosis or therapy might qualify.
This aspect is particularly relevant for innovators in broader technology fields. If a company is developing advanced sensing technology, AI-driven analytics, or even sophisticated drone platforms for data collection, they must be acutely aware of where their technology intersects with healthcare. A simple environmental sensor deployed via an autonomous system might be purely a tech innovation. However, if that sensor is adapted to monitor a patient’s room temperature for comfort (a basic function) or to remind them to take medication (a low-risk informational function), it might then fall under Class I medical device regulation. The crucial distinction lies in the intended use and the claims made about the product’s function. Innovators must meticulously define their product’s purpose to accurately determine its regulatory class and navigate the pathway efficiently, recognizing that even subtle shifts in intended use can move a product between classifications, demanding vastly different regulatory burdens.
Impact on Technological Advancement and Public Trust
The existence of the Class I medical device category is not merely a bureaucratic detail; it plays a vital role in shaping the pace and direction of technological advancement, particularly in how quickly safe, beneficial innovations can reach the public. It strikes a delicate balance between encouraging innovation and safeguarding public health.
Fostering Responsible Innovation
By creating a lower-barrier entry point for low-risk devices, the Class I classification actively fosters innovation. It allows smaller entities, academic researchers, and startups to experiment with and bring to market simple yet effective tools without the prohibitive costs and timeframes associated with higher-risk classifications. This encourages a broader spectrum of creativity and problem-solving, leading to a wider array of useful products that improve patient care, enhance daily living, or support healthcare infrastructure in non-critical ways. For instance, innovative designs for crutches, redesigned simple examination lights, or novel packaging for sterile dressings can all emerge faster and more efficiently thanks to this tiered system.
Furthermore, this framework instills a culture of responsible innovation. Even with reduced regulatory scrutiny, manufacturers of Class I devices are still bound by general controls. This means they must implement quality management systems, label their products accurately, report adverse events, and ensure their products are manufactured under appropriate conditions. This foundational level of accountability ensures that even the simplest technologies meet basic safety and efficacy standards, thereby building and maintaining public trust in medical technology as a whole. Innovators in adjacent fields, such as those developing components for autonomous delivery systems or advanced environmental sensors, must appreciate that if their technology ever veers into a medical application, this baseline of responsibility will apply.
The Evolving Regulatory Frontier for New Technologies
The regulatory landscape is constantly challenged by the rapid pace of technological advancement. Emerging technologies like artificial intelligence (AI), advanced robotics, the Internet of Things (IoT), and sophisticated sensors are blurring traditional boundaries, demanding that regulators adapt their frameworks. While many of these cutting-edge innovations will fall into higher-risk categories if they perform diagnostic or therapeutic functions, understanding Class I remains critical.
Consider a future where autonomous systems are extensively used for logistics in healthcare. While the drone itself might not be a medical device, a simple, non-diagnostic sensor it carries to monitor air quality in a sterile environment, or a basic temperature indicator for transported samples, could potentially be classified as Class I. Similarly, AI algorithms that merely organize information for a physician, without interpreting data for diagnostic purposes, might also find themselves in this lower-risk class. The key for innovators in these fields is to delineate the intended use precisely. By doing so, they can strategically design their products or components to fall into Class I where appropriate, leveraging the faster market access this classification offers, while still maintaining compliance and upholding safety standards. This foresight is a hallmark of sophisticated technological development, recognizing that innovation isn’t just about creating new tech, but also about understanding its rightful place within societal and regulatory structures.
Future Perspectives: Class I Devices in an Integrated Tech Ecosystem
As technology continues to intertwine with healthcare, Class I medical devices, despite their low-risk profile, are poised to play an increasingly integral role in the broader ecosystem of health tech and innovation. Their simplicity and relative ease of market entry make them ideal candidates for rapid integration and iterative development within complex systems.
Interoperability and Low-Risk Health Solutions
The future of healthcare technology is inherently interconnected. Advanced health systems will rely on seamless data flow, interoperability between various devices, and integrated platforms. In this context, Class I devices can serve as critical, low-friction components that facilitate data capture or basic patient support within larger, more sophisticated frameworks. Imagine a scenario where a basic, Class I compliant sensor is embedded into a smart home system to monitor a specific environmental parameter for an elderly patient. Or perhaps a simple, non-diagnostic software module that facilitates the organization of patient reminders, seamlessly interacting with an AI-driven personal assistant. These simple, safe elements, individually classified as Class I, can contribute significantly to the overall effectiveness and user-friendliness of complex health ecosystems without introducing substantial new regulatory hurdles at every integration point.
For innovators focusing on larger technological architectures, understanding how Class I devices fit into this puzzle is essential. It enables them to design solutions that incorporate simple, well-understood components for straightforward functions, reserving higher-level regulatory scrutiny for the truly high-risk diagnostic or therapeutic elements. This modular approach to compliance can accelerate development cycles for integrated health solutions, allowing innovators to focus their most intensive regulatory efforts on the critical, high-impact technologies while still ensuring foundational safety across the entire system.
The Role of Simple, Safe Components in Complex Medical Systems
The continuous advancement in sensor technology, miniaturization, and data processing means that even seemingly simple functions can be delivered with unprecedented precision and efficiency. A basic thermometer, a Class I device, can now be wireless, connected, and integrated into a remote monitoring platform. While the platform itself or any diagnostic algorithms it employs might be higher class, the individual low-risk components retain their classification, benefiting from a less burdensome path to market.
This principle extends to various domains of technology and innovation. Developers working on advanced robotics might create simple, physical interfaces for patient interaction that are Class I. Companies in remote sensing and data analytics might offer basic data visualization tools that fall into this category. The key takeaway for any innovator is that the regulatory classification system, particularly Class I, provides a clear pathway for introducing useful, low-risk tools into the medical domain. By carefully defining intended use and adhering to general controls, technology companies can contribute to healthcare innovation, leveraging their expertise in areas like advanced materials, manufacturing, and digital interfaces, even if their core business isn’t directly medical. This approach broadens the pool of innovators contributing to healthcare, leading to a more diverse, dynamic, and ultimately, more effective health tech landscape.