Understanding the Biologics License Application (BLA) Process
The journey of a novel biological product from laboratory discovery to patient access is a complex and rigorously regulated undertaking. Central to this process in the United States is the Biologics License Application (BLA). A BLA is the comprehensive submission required by the Food and Drug Administration (FDA) to gain approval to market and sell a biological product. Unlike the New Drug Application (NDA) for small-molecule drugs, a BLA is specifically designed for products derived from living organisms, encompassing a broad spectrum of therapeutic agents, vaccines, diagnostics, and other biological materials. Understanding the intricacies of the BLA is paramount for any company aiming to bring these life-saving and life-enhancing innovations to the public.
The Regulatory Framework: Ensuring Safety and Efficacy
The regulation of biological products is rooted in the Public Health Service Act (PHSA) and is overseen by the FDA’s Center for Biologics Evaluation and Research (CBER). The core objective of the BLA process is to ensure that a biological product is safe and effective for its intended use, and that it is manufactured consistently to meet high quality standards. This involves a multi-stage review process that scrutinizes every aspect of the product, from its fundamental scientific basis to its manufacturing controls and proposed labeling. The BLA serves as the definitive document where all this critical information is compiled and presented to the FDA for review. It is not merely a formality but a substantive scientific and regulatory submission that underpins public trust in biological medicines.
The BLA process is designed to be exhaustive, demanding extensive data and documentation. This includes detailed information about the product’s chemistry, manufacturing, and controls (CMC), as well as preclinical and clinical studies that demonstrate its safety and efficacy. The FDA’s review is conducted by a team of experts in various scientific and medical disciplines, ensuring a thorough and impartial evaluation. The goal is not to hinder innovation but to safeguard public health by ensuring that only the most rigorously tested and reliably manufactured biological products reach the market.
Key Components of a Biologics License Application
A well-prepared BLA is a testament to the extensive research, development, and quality control measures undertaken by a sponsor. It is a comprehensive package of information designed to provide the FDA with a complete understanding of the biological product. While the specific requirements can vary depending on the type of biologic, several core components are consistently expected. These components collectively form the backbone of the BLA, enabling regulators to make informed decisions about product approval.
Chemistry, Manufacturing, and Controls (CMC)
The CMC section is arguably the most critical and often the most extensive part of a BLA. It details every aspect of how the biological product is made and how its quality is maintained. This includes:
- Manufacturing Process: A thorough description of the manufacturing process, from the sourcing of raw materials to the final purification and filling of the product. This involves outlining the cell lines or microbial strains used, the media and reagents employed, and the various steps involved in production, such as fermentation, cell culture, or purification. Any modifications or changes to the manufacturing process must be meticulously documented and justified.
- Characterization: Detailed information on the physicochemical and biological properties of the product. This involves using a variety of analytical techniques to identify and quantify the active ingredient, impurities, and other relevant components. The goal is to establish a clear profile of the product and to ensure its identity, purity, potency, and strength.
- Analytical Methods: A description of the validated analytical methods used to test the product at various stages of manufacturing and for the final release. These methods must be robust, sensitive, and specific to accurately assess the quality attributes of the biologic.
- Stability Studies: Data demonstrating the product’s stability under various storage conditions (temperature, humidity, light) over time. This information is crucial for determining the product’s shelf life and recommended storage conditions, ensuring that it remains safe and effective until its expiration date.
- Facility Information: Details about the manufacturing facilities, including their design, equipment, and quality systems, to ensure they meet current Good Manufacturing Practices (cGMP) standards. This includes information on environmental controls, personnel qualifications, and validation of equipment and processes.
Nonclinical Studies
This section presents the results of laboratory and animal studies conducted to assess the safety and biological activity of the product before human testing. These studies help to establish a scientific rationale for the intended use and to identify potential toxicities.
- Pharmacology: Studies that investigate how the biologic interacts with its biological targets and produces its intended effects. This can include in vitro and in vivo studies to understand the mechanism of action.
- Pharmacokinetics (PK): Studies that examine how the body absorbs, distributes, metabolizes, and excretes the biological product. This provides insights into dosing regimens and potential accumulation.
- Toxicology: Studies designed to identify potential adverse effects of the biologic. This can include acute, subchronic, and chronic toxicity studies, as well as reproductive and developmental toxicity studies, genotoxicity, and carcinogenicity studies where appropriate.
Clinical Studies
This is where the safety and efficacy of the biological product are evaluated in humans. Clinical studies are typically conducted in phases, with each phase building upon the data from the previous one.
- Phase 1: Small studies, often involving healthy volunteers, to assess safety, determine a safe dosage range, and identify side effects.
- Phase 2: Larger studies involving patients with the target condition to evaluate efficacy and further assess safety.
- Phase 3: Large-scale, randomized controlled trials involving a diverse patient population to confirm efficacy, monitor side effects, compare it to commonly used treatments, and collect information that will allow the biologic to be used safely.
- Clinical Data Analysis: A comprehensive analysis of all clinical trial data, including statistical evaluations, to demonstrate the product’s benefit-risk profile.
Labeling

The proposed labeling for the biological product is a crucial component of the BLA. This includes the package insert (prescribing information), patient information leaflets, and any other materials intended to communicate essential information to healthcare professionals and patients.
- Prescribing Information: This detailed document provides healthcare providers with comprehensive information on the product’s indications, dosage, administration, contraindications, warnings, precautions, adverse reactions, drug interactions, and clinical pharmacology.
- Patient Labeling: Information designed for patients, explaining the product’s benefits, risks, and how to use it safely.
- Proposed Marketing Materials: While not always a formal part of the initial BLA, the FDA may review proposed marketing materials to ensure they are consistent with the approved labeling and do not make unsubstantiated claims.
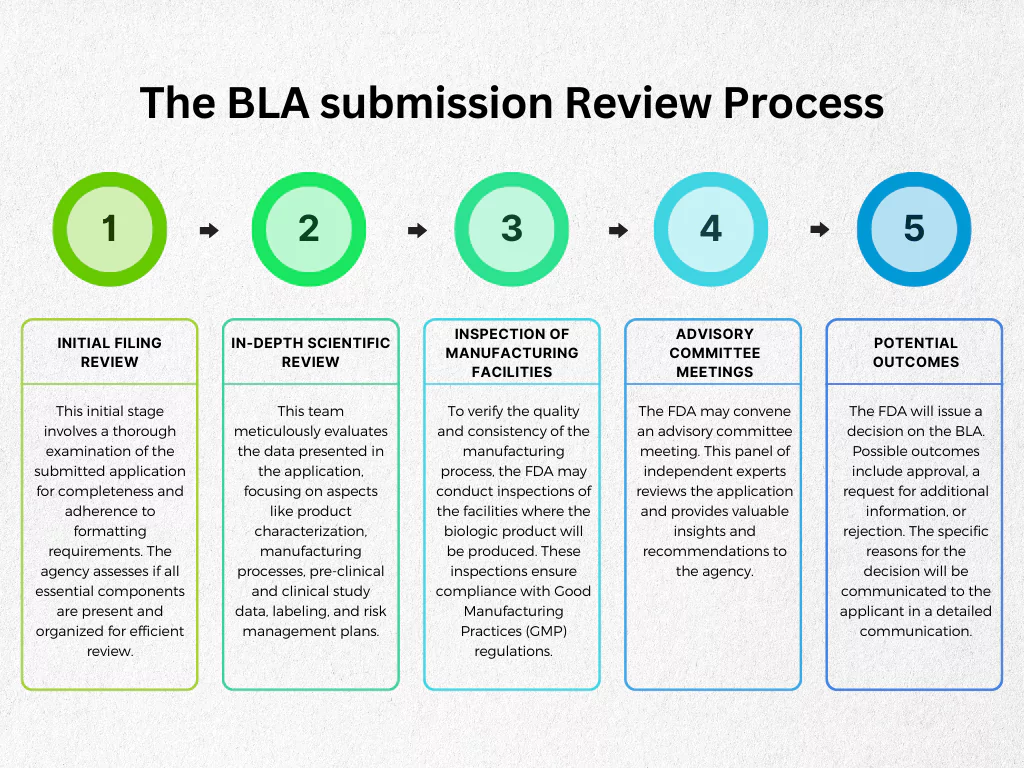
The BLA Review Process: A Rigorous Evaluation
Submitting a BLA is just the first step. The FDA then embarks on a thorough review process to assess the application’s completeness and the scientific merits of the data presented. This process is designed to be transparent and collaborative, with opportunities for dialogue between the applicant and the regulatory agency.
Initial Review and Filing Decision
Upon submission, the FDA conducts an initial review to determine if the BLA is complete and acceptable for filing. This “filing review” typically takes about 60 days. If the application is deemed complete, it is officially filed, and the substantive review begins. If it is deemed incomplete, the FDA will issue a “Refuse to Receive” letter, outlining the deficiencies that need to be addressed before the application can be resubmitted.
Substantive Review
The core of the BLA review involves a detailed scientific and regulatory evaluation by a multidisciplinary team of FDA experts. This team includes physicians, statisticians, chemists, pharmacologists, and other specialists relevant to the specific biological product.
- Review Teams: Dedicated teams are assigned to each BLA, responsible for evaluating all aspects of the submission.
- Information Requests: During the review, the FDA may issue Information Requests (IRs) to the applicant for clarification or additional data. These requests are critical for addressing any ambiguities or concerns identified by the reviewers.
- Advisory Committee Meetings: For certain complex or novel products, the FDA may convene an advisory committee meeting. This committee, comprised of external experts, provides independent advice to the FDA on specific scientific and medical issues related to the BLA.
Facility Inspections
As part of the BLA review, the FDA conducts pre-approval inspections of the manufacturing facilities. These inspections are crucial to verify that the manufacturing processes described in the BLA are being followed and that the facilities comply with cGMP regulations.
- On-Site Inspections: FDA inspectors visit the manufacturing sites to assess the quality systems, equipment, personnel, and overall operational compliance.
- Data Verification: Inspectors may review batch records, quality control data, and other documentation to ensure the consistency and reliability of the manufacturing process.
Decision and Post-Approval Obligations
Following the comprehensive review and facility inspections, the FDA makes a decision on the BLA.
- Approval: If the FDA determines that the biological product is safe and effective for its intended use, and that it can be manufactured consistently to meet quality standards, it will issue an approval letter.
- Complete Response Letter (CRL): If the FDA identifies deficiencies that prevent approval, it will issue a Complete Response Letter. This letter details the issues that must be addressed before the BLA can be reconsidered. This may involve conducting additional studies, providing more data, or making changes to the manufacturing process.
- Post-Approval Monitoring: Even after approval, the FDA continues to monitor the safety and efficacy of biological products through post-market surveillance, adverse event reporting, and potential post-approval studies or commitments. Manufacturers also have ongoing obligations to report manufacturing changes and any new safety information.

The Significance of the BLA for Biologic Innovation
The Biologics License Application is more than just a regulatory hurdle; it is a cornerstone of innovation in the field of biological medicine. By establishing a rigorous framework for review and approval, the BLA process ensures that groundbreaking scientific discoveries can be translated into safe and effective therapies for patients. It fosters an environment where investment in research and development is encouraged, knowing that a clear pathway exists for bringing valuable biological products to market.
The BLA process, while demanding, serves a vital public health purpose. It provides a critical assurance to healthcare providers and patients that biological products have undergone thorough scientific scrutiny and meet stringent quality standards. This confidence is essential for the widespread adoption and therapeutic success of these complex and often life-saving treatments. Ultimately, the BLA is a critical mechanism that balances the drive for innovation with the imperative of public safety, ensuring that the benefits of biological advancements are realized responsibly.