Understanding Hemoglobin A2 in the Context of Blood Health
Hemoglobin, the protein responsible for carrying oxygen in red blood cells, is a critical component of our circulatory system. While most individuals are familiar with the primary form of hemoglobin, Hemoglobin A (HbA), the human body also produces other forms, including Hemoglobin A2 (Hgb A2). Understanding Hgb A2 is vital for a comprehensive grasp of blood health and the diagnosis of certain genetic blood disorders. This article delves into the nature of Hgb A2, its significance, how it is measured, and its implications for health.
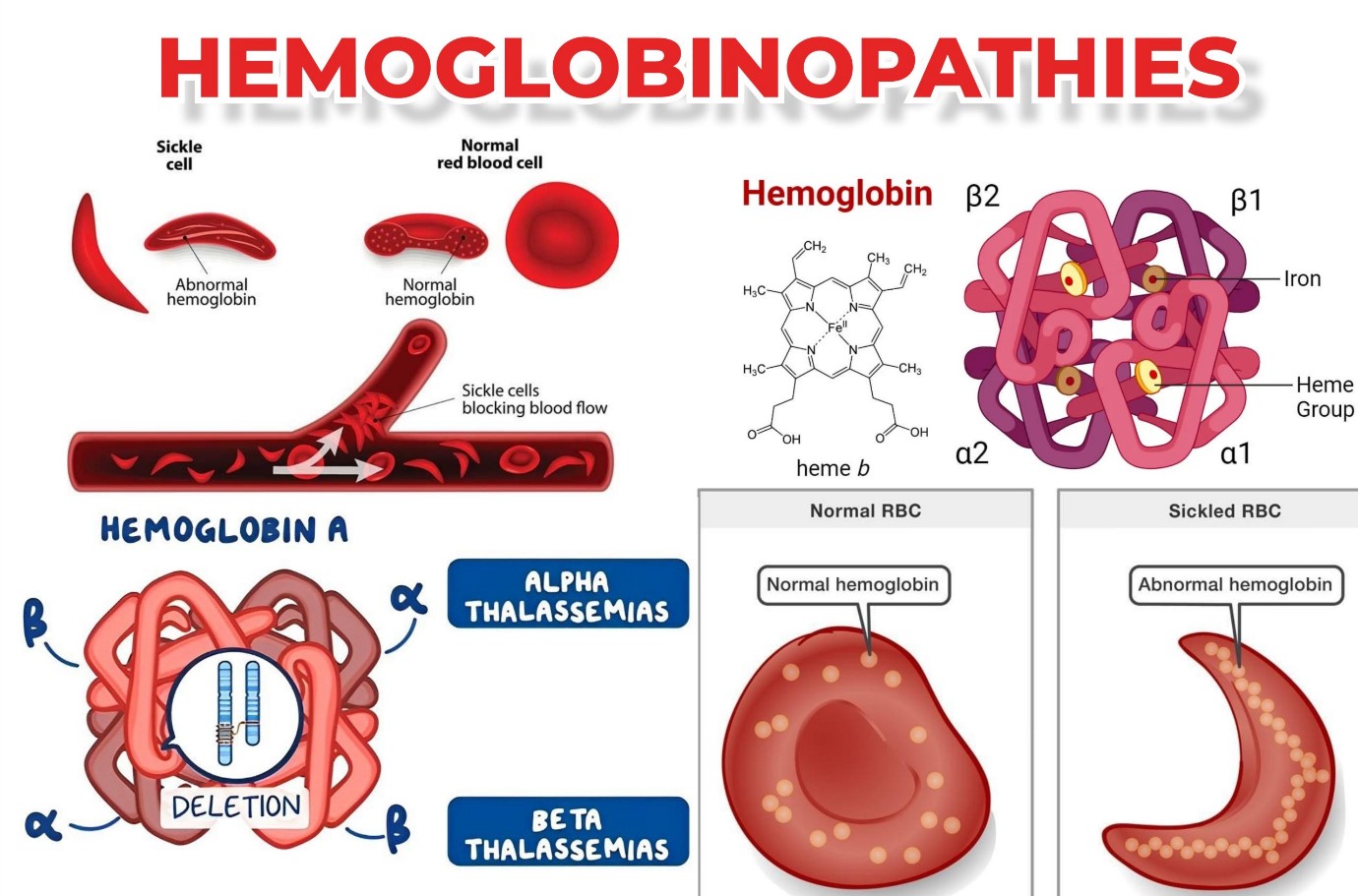
The Molecular Landscape of Hemoglobin
To comprehend Hgb A2, it’s essential to first understand the structure of hemoglobin itself. Hemoglobin is a tetramer, meaning it is composed of four protein subunits. Each subunit contains a heme group, which is a ring-like structure that binds to an iron atom. It is this iron atom that directly interacts with oxygen molecules.
In adults, the most common type of hemoglobin is Hemoglobin A (HbA). HbA consists of two alpha-globin chains and two beta-globin chains, denoted as α₂β₂. This structure allows for efficient oxygen transport from the lungs to the tissues and carbon dioxide transport back to the lungs.
However, as the body matures, there are shifts in the production of globin chains. In fetuses, Hemoglobin F (HbF) is dominant, composed of two alpha-globin chains and two gamma-globin chains (α₂γ₂). HbF has a higher affinity for oxygen than HbA, which is crucial for transferring oxygen from the maternal circulation to the fetus in the placenta. After birth, HbF production gradually decreases, and HbA becomes the predominant form.
The Composition and Origin of Hemoglobin A2
Hemoglobin A2 (Hgb A2) is a minor adult hemoglobin variant. Unlike HbA and HbF, Hgb A2 is characterized by its unique combination of globin chains. It consists of two alpha-globin chains and two delta-globin chains, represented as α₂δ₂. The delta-globin gene (HBD) is located on the same chromosome as the beta-globin gene (HBB), with both being part of the beta-globin gene cluster.
The production of Hgb A2 begins during the later stages of fetal development and continues into adulthood, albeit at a much lower level than HbA. In healthy individuals, Hgb A2 typically constitutes about 1.5% to 3.5% of the total hemoglobin in adults. This relatively small percentage belies its significant clinical importance.
The regulation of delta-globin gene expression is complex and influenced by various factors, including transcriptional regulators and epigenetic modifications. While the exact physiological role of Hgb A2 remains a subject of ongoing research, it is understood to play a role in oxygen transport, though its oxygen-binding affinity is slightly lower than that of HbA.
Measuring Hemoglobin A2: Diagnostic Techniques
The accurate quantification of Hgb A2 levels is primarily achieved through laboratory analysis of a blood sample. Several techniques are employed for this purpose, each offering varying degrees of precision and accessibility.
1. High-Performance Liquid Chromatography (HPLC)
HPLC is currently the gold standard for measuring Hgb A2. This method separates hemoglobin variants based on their differing physical and chemical properties, such as their charge and hydrophobicity. A blood sample is processed, and the hemolysate (red blood cells broken open to release hemoglobin) is injected into the HPLC system. As the hemolysate passes through a column packed with a stationary phase, different hemoglobin types elute at different times, allowing for their identification and quantification. HPLC offers high sensitivity and specificity, making it the preferred method for routine clinical testing and research.
2. Capillary Electrophoresis (CE)
Capillary electrophoresis is another highly accurate method for hemoglobin analysis. In CE, charged molecules migrate through a narrow capillary filled with an electrolyte solution under the influence of an electric field. Hemoglobin variants separate based on their charge and size. Similar to HPLC, CE can effectively resolve and quantify Hgb A2 and other hemoglobin types. It is known for its speed and minimal sample requirement.
3. Acid Elution or Alkaline Gel Electrophoresis
Older methods, such as acid elution and alkaline gel electrophoresis, were historically used for hemoglobin analysis. While still capable of detecting certain hemoglobinopathies, these techniques are less precise for quantifying Hgb A2 compared to HPLC or CE. They are often used as initial screening tests or in settings where more advanced equipment is unavailable. Alkaline gel electrophoresis separates hemoglobins based on their charge at an alkaline pH, while acid elution can help identify red blood cells with abnormal hemoglobin content.
4. Spectrophotometry and Other Immunoassays
While less common for direct Hgb A2 quantification, some colorimetric assays or immunoassays might be used in specific research or diagnostic contexts. However, these methods generally lack the resolution and accuracy needed for precise Hgb A2 measurement compared to chromatographic or electrophoretic techniques.
The choice of measurement technique often depends on the laboratory’s resources, the specific diagnostic question, and established protocols. Regular calibration and quality control are essential to ensure the accuracy of Hgb A2 measurements.
The Clinical Significance of Hemoglobin A2 Levels

The measurement of Hgb A2 levels is particularly crucial in the diagnosis and management of beta-thalassemia trait and beta-thalassemia major. Thalassemia is a group of inherited blood disorders characterized by reduced or absent synthesis of globin chains, leading to reduced hemoglobin production and anemia.
1. Beta-Thalassemia Trait (Minor)
In individuals with beta-thalassemia trait, they inherit one normal beta-globin gene and one mutated beta-globin gene. This typically results in a mild form of anemia or no anemia at all, often discovered incidentally during routine blood tests. A key diagnostic marker for beta-thalassemia trait is an elevated level of Hgb A2. In these individuals, the body attempts to compensate for the reduced production of beta-globin chains by increasing the production of delta-globin chains, leading to a higher proportion of Hgb A2. Typically, Hgb A2 levels in beta-thalassemia trait range from 3.5% to 7% or higher. This elevation is a sensitive indicator and helps differentiate beta-thalassemia trait from iron deficiency anemia, which is a more common cause of microcytic anemia.
2. Beta-Thalassemia Major
Beta-thalassemia major, also known as Cooley’s anemia, is a severe form of the disease resulting from the inheritance of two mutated beta-globin genes. Individuals with beta-thalassemia major have very little or no production of beta-globin chains, leading to severe anemia that typically manifests in infancy. In these cases, while Hgb A2 levels can sometimes be elevated, the overall hemoglobin picture is dominated by the presence of significant amounts of fetal hemoglobin (HbF) as the body attempts to compensate. The diagnosis of beta-thalassemia major relies on a combination of clinical presentation, red blood cell indices, and detailed hemoglobin electrophoresis showing a profound lack of HbA and the presence of HbF.
3. Alpha-Thalassemia
While Hgb A2 is primarily associated with beta-thalassemia, it’s worth noting that alpha-thalassemia, which involves reduced production of alpha-globin chains, has a different diagnostic profile. In alpha-thalassemia, the levels of Hgb A2 are usually normal, and the diagnostic focus is on the presence of abnormal hemoglobins like Hemoglobin H (HbH) and Hemoglobin Barts.
4. Other Hemoglobinopathies
Beyond thalassemia, certain rare hemoglobinopathies might influence Hgb A2 levels. For example, delta-beta-thalassemia, a condition where there is a deletion or defect in both the delta and beta-globin genes, can lead to a complete absence of Hgb A2. In contrast, hereditary persistence of fetal hemoglobin (HPFH) can sometimes be associated with normal or slightly elevated Hgb A2, depending on the specific genetic defect.
5. Monitoring and Management
For individuals diagnosed with beta-thalassemia trait, understanding their Hgb A2 levels is important for genetic counseling. If both partners carry the beta-thalassemia trait, they have a 25% chance of having a child with beta-thalassemia major. Monitoring Hgb A2 in pregnant women or couples planning a family can help identify at-risk pregnancies. For those with beta-thalassemia major, ongoing monitoring of hemoglobin levels, including the proportions of different hemoglobin types, is crucial for guiding treatment regimens such as blood transfusions and iron chelation therapy.
Factors Influencing Hemoglobin A2 Levels
While the genetic basis of beta-thalassemia trait is the most significant factor affecting Hgb A2 levels, other factors can sometimes influence its measurement or interpretation.
1. Vitamin B12 and Folate Deficiency
Severe deficiencies in Vitamin B12 and folate can lead to megaloblastic anemia. In some instances, these deficiencies can also affect globin chain synthesis and, consequently, the proportion of Hgb A2. However, the effect is generally less pronounced and consistent compared to beta-thalassemia trait. If a patient presents with anemia and an elevated Hgb A2, it is crucial to rule out co-existing iron deficiency and megaloblastic anemia.
2. Pregnancy
Hgb A2 levels can sometimes show slight variations during pregnancy. While typically not diagnostic, significant deviations might warrant further investigation.
3. Myeloproliferative Disorders
In rare cases, certain myeloproliferative disorders, which affect the bone marrow’s production of blood cells, can lead to alterations in hemoglobin composition, potentially impacting Hgb A2 levels.
4. Artifacts and Interference
As with any laboratory test, artifacts or interference can occur. These can arise from issues with sample collection, processing, or the analytical method itself. Therefore, adherence to strict laboratory protocols and quality control measures is paramount for accurate Hgb A2 quantification.

Conclusion: The Small Molecule with a Big Impact
Hemoglobin A2, though a minor component of adult hemoglobin, plays a pivotal role in hematology. Its accurate measurement is indispensable for the diagnosis of beta-thalassemia trait, a common inherited blood disorder. By understanding the molecular basis of Hgb A2 and the techniques used to quantify it, healthcare professionals can effectively identify individuals at risk, provide genetic counseling, and manage affected patients. The consistent elevation of Hgb A2 in beta-thalassemia trait serves as a clear indicator, allowing for timely intervention and improved outcomes for those affected by hemoglobinopathies. As research continues, a deeper understanding of Hgb A2’s physiological function may further illuminate its role in overall blood health.