Sanfilippo syndrome, a rare genetic disorder, represents a formidable challenge in the realm of pediatric medicine and genetic research. While the title itself might seem obscure to many, understanding its core mechanisms, clinical manifestations, and the ongoing scientific endeavors to combat it is crucial for raising awareness and fostering hope. This condition, primarily affecting children, disrupts the body’s ability to break down complex sugar molecules, leading to a cascade of severe developmental and physical consequences. At its heart, Sanfilippo syndrome is a story of cellular machinery gone awry, impacting the very building blocks of life and progressively eroding a child’s cognitive and physical abilities.

Understanding the Genetic Basis of Sanfilippo Syndrome
Sanfilippo syndrome is fundamentally a lysosomal storage disorder, a group of inherited metabolic diseases characterized by the deficiency of specific enzymes. These enzymes are vital for breaking down complex sugar molecules (glycosaminoglycans, or GAGs) within the cell’s lysosomes – the cellular “recycling centers.” When these enzymes are absent or dysfunctional, GAGs accumulate within the lysosomes, leading to cellular damage and dysfunction, particularly in tissues with high metabolic activity like the brain.
The Role of Lysosomes and Glycosaminoglycans
Lysosomes contain a variety of enzymes responsible for degrading waste products and cellular debris. Among these are enzymes crucial for breaking down long chains of sugar molecules known as GAGs. GAGs, also called mucopolysaccharides, are essential components of connective tissues, bone, cartilage, and cell surfaces. In a healthy individual, GAGs are continuously synthesized and degraded. However, in Sanfilippo syndrome, a defect in the genes responsible for producing specific enzymes involved in GAG degradation prevents this process from occurring efficiently.
The most common GAGs that accumulate in Sanfilippo syndrome are heparan sulfate and, to a lesser extent, dermatan sulfate and chondroitin sulfate. These molecules are not fully broken down into their constituent parts and instead build up within the lysosomes of various cells throughout the body. This accumulation exerts pressure on the lysosome, eventually causing it to rupture and release its contents, leading to cellular damage and death. The progressive nature of this accumulation is what drives the devastating symptoms of the syndrome.
The Different Types of Sanfilippo Syndrome
Sanfilippo syndrome is not a single entity but rather a group of related disorders, classified into four main subtypes based on the specific enzyme deficiency. These subtypes are designated by letters A, B, C, and D. Each subtype is caused by a mutation in a different gene, each responsible for encoding a crucial enzyme in the GAG degradation pathway.
-
Sanfilippo Syndrome Type A (MPS III A): This is the most common and often the most severe form. It is caused by a deficiency in the enzyme sulfamidase, encoded by the SGSH gene. This deficiency leads to a rapid accumulation of heparan sulfate. Children with Type A often present with early and significant neurological symptoms.
-
Sanfilippo Syndrome Type B (MPS III B): This subtype arises from a deficiency in the enzyme alpha-N-acetylglucosaminidase, encoded by the NAGLU gene. While still severe, Type B can sometimes progress slightly slower than Type A.
-
Sanfilippo Syndrome Type C (MPS III C): Caused by a deficiency in the enzyme acetyl-CoA:alpha-glucosaminide N-acetyltransferase, encoded by the HGSNAT gene, this type is relatively rare. The primary defect in Type C is a problem with the initial step of heparan sulfate breakdown.
-
Sanfilippo Syndrome Type D (MPS III D): This subtype results from a deficiency in the enzyme N-acetylglucosamine-6-sulfatase, encoded by the GNS gene. Like Type B, Type D can exhibit slightly slower progression compared to Type A.
Despite the differences in the specific enzyme affected, the overall clinical picture and the progressive nature of the disease are remarkably similar across all subtypes, making a definitive diagnosis and understanding of the underlying genetic cause critical for management and research.
Clinical Manifestations: The Progressive Impact of Sanfilippo Syndrome
The insidious onset and relentless progression of Sanfilippo syndrome are its most challenging aspects. Symptoms typically begin to emerge in early childhood, between the ages of 2 and 6, and worsen over time, impacting nearly every aspect of a child’s development and well-being. The hallmark of the disease is the progressive neurodegeneration, but other organ systems are also affected.
Early Childhood Development and Behavioral Changes
In the early stages, children with Sanfilippo syndrome may appear to develop normally, which can mask the underlying pathology. However, subtle delays in speech and language development, gross and fine motor skills, and social interaction can begin to surface. One of the most striking early features is often a significant behavioral component. Children may exhibit hyperactivity, impulsivity, aggression, and difficulty sleeping. These behavioral challenges can be exhausting for caregivers and are often among the first clear indicators that something is amiss.
As the disease progresses, the neurological impact becomes more pronounced. The accumulation of GAGs in the brain leads to neuronal damage and dysfunction. This results in a gradual but irreversible decline in cognitive abilities. Children lose previously acquired skills, such as language, problem-solving, and reasoning. The ability to learn new information diminishes, and memory impairment becomes evident. This cognitive regression is a core feature that distinguishes Sanfilippo syndrome from other developmental disorders.
Physical and Systemic Involvement
Beyond the brain, the widespread accumulation of GAGs affects various other organ systems, leading to a range of physical symptoms.
-
Skeletal Abnormalities: Bone and joint problems are common. Children may experience stiff joints, contractures (limited range of motion), and skeletal deformities such as kyphosis (a rounded upper back) or genu valgum (knock knees). The bones can become more fragile, increasing the risk of fractures.
-
Facial Features: While not as pronounced as in some other mucopolysaccharidoses, certain coarse facial features can develop over time, including a thickened appearance of the eyebrows, a broad nasal bridge, and enlarged tonsils and adenoids, which can contribute to breathing difficulties and sleep apnea.
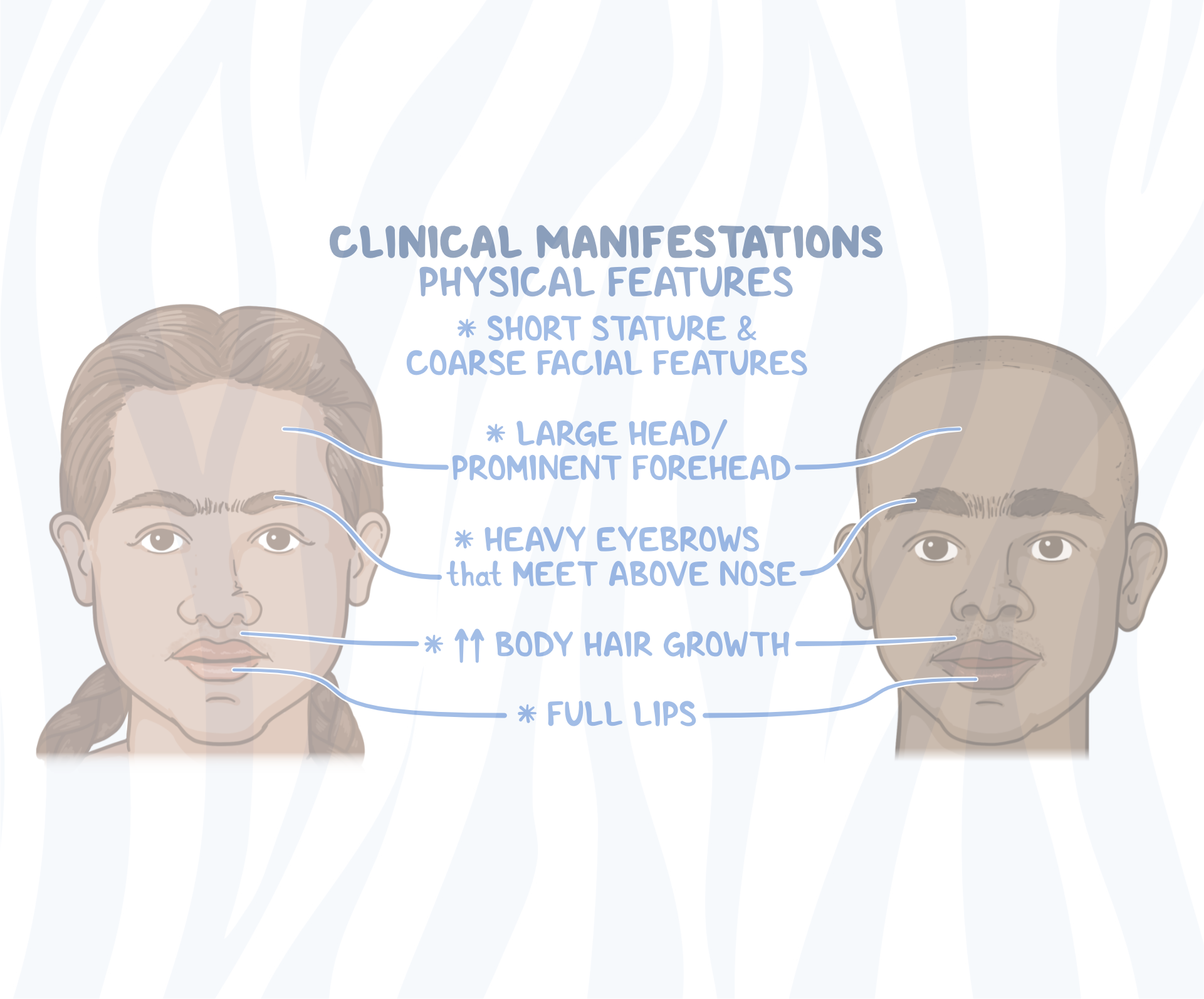
-
Cardiovascular Complications: The heart valves can be affected by GAG deposition, leading to valvular heart disease and progressive cardiac dysfunction. Regular cardiac monitoring is essential.
-
Respiratory Issues: Enlarged tonsils and adenoids, as mentioned, can cause airway obstruction. Furthermore, GAG accumulation in the respiratory tract can lead to chronic respiratory infections and decreased lung function.
-
Gastrointestinal Problems: Digestive issues such as diarrhea, constipation, and abdominal distension can also occur.
-
Vision and Hearing: Progressive vision impairment, including corneal clouding, and hearing loss are also frequently observed as the disease advances.
The combination of severe intellectual disability, progressive loss of function, and challenging behaviors makes Sanfilippo syndrome a profoundly debilitating condition that significantly impacts the lifespan and quality of life for affected children.
Diagnosis and Therapeutic Approaches: The Pursuit of Effective Interventions
The diagnosis of Sanfilippo syndrome, while challenging due to its rarity and the subtlety of early symptoms, is crucial for initiating supportive care and participating in research endeavors. The current therapeutic landscape is primarily focused on managing symptoms and improving quality of life, as definitive cures remain elusive. However, significant scientific progress is being made in the development of novel treatment strategies.
Diagnostic Pathways for Sanfilippo Syndrome
Confirming a diagnosis of Sanfilippo syndrome involves a multi-faceted approach that begins with clinical suspicion based on the characteristic symptoms and developmental trajectory.
-
Biochemical Testing: The first step often involves biochemical analysis of urine. Elevated levels of heparan sulfate are a strong indicator of Sanfilippo syndrome. Specific assays can then be performed to quantify the precise levels of GAGs and identify patterns suggestive of particular subtypes.
-
Enzyme Assays: Once elevated GAGs are detected, specific enzyme assays are performed using blood or cultured cells. These tests measure the activity of the enzymes deficient in each Sanfilippo subtype (sulfamidase for Type A, alpha-N-acetylglucosaminidase for Type B, etc.). A significantly reduced or absent enzyme activity confirms the diagnosis and identifies the specific subtype of Sanfilippo syndrome.
-
Genetic Testing: Definitive confirmation and detailed characterization of the genetic defect are achieved through genetic testing. This involves sequencing the genes associated with each subtype to identify the specific mutations responsible for the enzyme deficiency. Genetic testing is invaluable for confirming the diagnosis, providing genetic counseling to families, and facilitating enrollment in clinical trials.
Current Management and Supportive Care
As there is no cure for Sanfilippo syndrome, management focuses on alleviating symptoms, preventing complications, and maximizing a child’s comfort and quality of life. This requires a multidisciplinary approach involving various medical specialists.
-
Symptomatic Treatment: This includes managing behavioral issues with medications and therapies, addressing sleep disturbances, and providing physical, occupational, and speech therapy to support development and maintain function for as long as possible.
-
Nutritional Support: Ensuring adequate nutrition is vital, especially as swallowing difficulties can arise. Feeding tubes may be necessary in later stages.
-
Medical Interventions: Regular monitoring and treatment of specific complications are essential. This includes cardiac evaluations, respiratory support, management of skeletal issues, and addressing vision and hearing impairments.
-
Family Support and Education: Caring for a child with Sanfilippo syndrome is an immense undertaking. Comprehensive support and education for families are critical, providing them with resources, coping strategies, and access to support networks.

Emerging Therapeutic Frontiers: Gene Therapy and Beyond
The scientific community is actively engaged in developing disease-modifying therapies that aim to address the root cause of Sanfilippo syndrome. Gene therapy has emerged as a particularly promising avenue.
-
Gene Therapy: This approach seeks to deliver a functional copy of the deficient gene into the patient’s cells. By providing the correct genetic blueprint, the body can then produce the missing enzyme, thereby restoring the GAG degradation pathway. Several clinical trials using gene therapy for Sanfilippo syndrome are underway, with early results showing potential for slowing disease progression, particularly in the neurological domain. These trials typically involve delivering the therapeutic gene via a viral vector, often targeting cells in the brain.
-
Enzyme Replacement Therapy (ERT): While ERT has been successful for some other lysosomal storage disorders, its effectiveness for Sanfilippo syndrome is limited. The primary challenge is the blood-brain barrier, which prevents many systemically administered enzymes from reaching the central nervous system, where the most devastating effects occur.
-
Substrate Reduction Therapy (SRT): This strategy aims to reduce the production of GAGs, thereby lessening the burden on the impaired degradation pathway. While still in early stages of development for Sanfilippo syndrome, SRT represents another potential therapeutic avenue.
The ongoing research and development in these areas offer a beacon of hope for families affected by Sanfilippo syndrome, signifying a determined global effort to unravel the complexities of this rare disease and ultimately find effective treatments.